Integrals
The integrals module (namespace lible::ints) provides utilities to calculate molecular
integrals over Gaussian-type basis functions. The implementation is based on the
McMurchie-Davidson scheme; calculation of the
two-electron integrals makes use of the SHARK algorithm.
Some of the notations and conventions here might bear a close resemblance to what is in the
“Molecular Electronic-Structure Theory”.
This book served as the main reference in implementing the library. In order to use the library,
include the main header <lible/ints/ints.hpp> in your source code.
Definitions & conventions
Lible provides utilities to calculate molecular integrals over contracted Gaussian type atomic orbitals (GTAOs). A contracted GTAO can be written as
where \(g^{lm}_{\mu,i}\) denotes a primitive Gaussian basis function. The contraction depth is denoted by \(K\) and the contraction coefficients by \(d_{\mu,i}\). The normalization coefficient \(N_{\mu}\) is usually obtained from the overlap integral
The function lible::ints::calcShellNorms may be used for that purpose. The primitive Gaussian
basis functions are normalized, and depend on the orbital angular momentum \(l\) and its
projection, \(m_l = -l, -l + 1, \ldots, l\). Omitting here the \(\mu\)-label, a primitive
Gaussian basis function can be written as
The quantities in this expression are defined as:
\(\mathbf{r} = (x, y, z)\) – an arbitrary point in space.
\(\mathbf{A} = (A_x, A_y, A_z)\) – point in space where the basis function is centered, i.e., a nucleus.
\(\mathbf{r}_A = (x - A_x, y - A_y, z - A_z)\).
\(\mathbf{a} = (a_1,\ldots,a_K)\) – list of Gaussian primitive exponents.
\(N_{l} (a_i)\) – pure (harmonic) Gaussian primitive normalization coefficient,
lible::ints::purePrimitiveNorm, that is calculated analytically from
\(S_{lm} (\mathbf{A})\) – a real-valued solic harmonic. The explicit expressions for these shall not be given here. Essentially, the solid harmonics can be expressed in terms of Cartesian directions through the transformation \(t_{lm;ijk}\),
\[S_{lm} = \sum_{ijk} t_{lm,ijk} x_A^i y_A^j z_A^k\]where the transformation coefficients are given by given by eq. (9.1.9) in “Molecular Electronic-Structure Theory”.
The Cartesian to spherical transformation is accessible from the library via the function
lible::ints::sphericalTrafo which returns the transformation coefficients up to \(l=9\).
Lible follows a convention where the spherical atomic orbitals are ordered alternatingly by the
\(m_l\) quantum number, as in the table:
l |
\(m_l\) |
|---|---|
0 |
0 |
1 |
0, 1,-1 |
2 |
0, 1,-1, 2,-2 |
3 |
0, 1,-1, 2,-2, 3,-3 |
The number of spherical harmonic atomic orbitals is given by \(N = 2l + 1\),
lible::ints::numSphericals.
The Cartesian Gaussian functions are ordered alphabetically by the Cartesian exponents. For the first few angular momenta, \(l = 0-3\), the Cartesian exponents are given by:
\(l\) |
Cartesian exponents |
|---|---|
0 |
1 |
1 |
(1, 0, 0), (0, 1, 0), (0, 0, 1) |
2 |
(2, 0, 0), (1, 1, 0), (1, 0, 1), (0, 2, 0), (0, 1, 1), (0, 0, 2) |
3 |
(3, 0, 0), (2, 1, 0), (2, 0, 1), (1, 2, 0), (1, 1, 1), (1, 0, 2), (0, 3, 0), (0, 2, 1), (0, 1, 2), (0, 0, 3) |
To get the Cartesian exponents for arbitrary angular momentum, l, use lible::ints::cartExps.
The total number of Cartesian exponents is given by \(N = (l + 1)(l + 2) / 2\),
lible::ints::numCartesians.
Available integral kernels
The library provides kernel functions for various integral types. A kernel function calculates the integrals for one batch, which is either one shell doublet, triplet or a quartet. In the spirit of the SHARK method, all of the kernel functions return the integrals normalized and in a spherical basis. The available integral kernels are summarized in the table below:
Integral |
Function |
|---|---|
\((a|b)\) |
|
\(\boldsymbol{\nabla}_{AB} (a|b)\) |
|
\((a|{-\tfrac{1}{2}}\nabla^2|b)\) |
|
\(\boldsymbol{\nabla}_{AB} (a|{-\tfrac{1}{2}}\nabla^2|b)\) |
|
\((a|\sum_q \frac{-q}{r_{1q}}|b)\) |
|
\((a|\sum_q \frac{-q \cdot \text{erf}(\omega r_{1q})}{r_{1q}}|b)\) |
|
\(\boldsymbol{\nabla}_{AB} (a|\sum_q \frac{-q}{r_{1q}}|b)\) |
|
\(\boldsymbol{\nabla}_{AB} (a|\sum_q \frac{-q \cdot \text{erf}(\omega r_{1q})}{r_{1q}}|b)\) |
|
\(\boldsymbol{\nabla}_{q} (a|\frac{-q}{r_{1q}}|b)\) |
|
\(\boldsymbol{\nabla}_{q} (a| \frac{-q \cdot \text{erf}(\omega r_{1q})}{r_{1q}}|b)\) |
|
\((a|\frac{-q}{r_{1q}}|b)\) |
|
\((a| \frac{-q \cdot \text{erf}(\omega r_{1q})}{r_{1q}}|b)\) |
|
\((a|\boldsymbol{\hat{\mu}}_O|b)\) |
|
\(\boldsymbol{\nabla}_{A} \times \boldsymbol{\nabla}_{B} (a|\sum_q \frac{-q}{r_{1q}}|b)\) |
|
\((a|{-\boldsymbol{\nabla}}|b)\) |
|
\((a|{-\boldsymbol{r} \times \boldsymbol{\nabla}}|b)\) |
|
\((a|\hat{p}_i \hat{V} \hat{p}_j|b)\) |
|
\((ab|\frac{1}{r_{12}}|cd)\) |
|
\((ab|\frac{1}{r_{12}}|c)\) |
|
\((a|\frac{1}{r_{12}}|b)\) |
|
\(\boldsymbol{\nabla}_{ABCD}(ab|\frac{1}{r_{12}}|cd)\) |
|
\(\boldsymbol{\nabla}_{ABC}(ab|\frac{1}{r_{12}}|c)\) |
|
\(\boldsymbol{\nabla}_{AB}(a|\frac{1}{r_{12}}|b)\) |
|
\((\boldsymbol{\nabla}_{AB} \otimes \boldsymbol{\nabla}_{AB})(a|\frac{1}{r_{12}}|b)\) |
|
\(\boldsymbol{\nabla}_{A} \times \boldsymbol{\nabla}_{B}(ab|\frac{1}{r_{12}}|cd)\) |
|
\(\boldsymbol{\nabla}_{A} \times \boldsymbol{\nabla}_{B}(ab|\frac{1}{r_{12}}|c)\) |
|
Main Interface
Typically, integral calculation in quantum chemistry programs involves choosing a molecular geometry
and a basis set. From this data, shells (lible::ints::Shell) can be constructed that contain
all the information required for calculating the integrals. In Lible, the shells are taken
to create a special data structure called the shell pair data (lible::ints::ShellPairData).
Therefore, typical integral calculation with Lible involves the shell pair data and specifying a
pair of shells with an index. Graphically, the flow of data from the initial geometry and basis set
to integral calculation can be illustrated as:
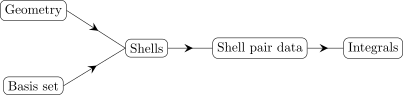
It should be noted here, that sometimes, the shells are transformed into the so-called shell data
(lible::ints::ShellData) data structure. This data structure can be used for calculating integrals
involving auxiliary basis functions, such as \((\mu\nu|P)\).
For convenience, it is possible to calculate some integrals directly, without utilizing the shell
pair data. Lible provides a special data structure that records all the information about geometry,
basis sets and shells for that purpose: lible::ints::Structure.
Using lible::ints::Structure, integrals can be calculated such that the management of shell pair
data is done under the hood. Graphically, this looks as follows:
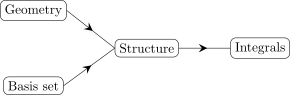
This approach can be preferable for testing and prototyping. For large scale implementation, the
previous scheme is probably preferable. For example, the function lible::ints::eri4 calculates
all of the four-center two-electron integrals. For large systems, the returned 4D array of integrals
would become too large to be stored in memory.
Let us assume that Lible is properly built/installed and linked against your code. To use the
library for calculating integrals, include the main header #include <lible/ints/ints.hpp> in
your source code. This header file constitutes the so-called main interface. The main interface
contains inclusions of some other Lible header files:

In the following we shall expose the contents of the main header file. After that, the documentation of the other header files will be provided.
<lible/ints/ints.hpp>
For the code snippets shown below, assume the following data is available:
std::string basis_set = "def2-svp";
std::string basis_set_aux = "def2-universal-jkfit";
std::vector<int> atomic_nrs_h2o{8, 1, 1};
std::vector<std::array<double, 3>> coords_h2o_ang{
{0.00000, 0.00000, 0.11779},
{0.00000, 0.75545,-0.47116},
{0.00000,-0.75545,-0.47116}};
-
vec2d overlap(const Structure &structure)
Calculates the overlap integrals. Uses OpenMP parallelization.
lible::ints::Structure structure(basis_set, atomic_nrs, coords_h2o_ang); lible::vec2d ovlp_ints = lible::ints::overlap(structure);
-
vec2d overlapKernel(size_t ipair, const ShellPairData &sp_data)
Calculates a batch of overlap integrals. The shell pair index
ipairrefers to a pair of shells in the shell pair datasp_data.for (size_t ipair = 0; ipair < sp_data.n_pairs_; ipair++) { // The index `ipair` is used by the kernel function to read the data necessary for // integral calculation. lible::vec2d ints_batch = lible::ints::overlapKernel(ipair, sp_data); ... }
-
std::array<vec2d, 6> overlapD1Kernel(size_t ipair, const ShellPairData &sp_data)
Calculates a batch of first derivative overlap integrals. Returns the derivative integrals for the atomic centers involved in the shell pair (
ipair): \(({\partial_A}_x, {\partial_A}_y, {\partial_A}_z, {\partial_B}_x, {\partial_B}_y, {\partial_B}_z)\).for (size_t ipair = 0; ipair < sp_data.n_pairs_; ipair++) { std::array<lible::vec2d, 6> ints_batch = lible::ints::overlapD1Kernel(ipair, sp_data); // The indices of the atoms (A and B) involved in the integrals batch can be obtained // from the shell pair data. size_t iatom_a = atomic_idxs_[2 * ipair]; size_t iatom_b = atomic_idxs_[2 * ipair + 1]; }
-
vec2d kineticEnergy(const Structure &structure)
Calculates the kinetic energy integrals. Uses OpenMP parallelization.
-
vec2d kineticEnergyKernel(size_t ipair, const ShellPairData &sp_data)
Calculates a batch of kinetic energy integrals.
-
std::array<vec2d, 6> kineticEnergyD1Kernel(size_t ipair, const ShellPairData &sp_data)
Calculates a batch of first derivative kinetic energy integrals.
-
vec2d nuclearAttraction(const Structure &structure)
Calculates nuclear attraction integrals. Uses OpenMP parallelization.
-
vec2d nuclearAttractionErf(const Structure &structure, const std::vector<double> &omegas)
Calculates the attenuated Coulomb attraction integrals. Uses OpenMP parallelization.
Important
The number of attenuation parameters
omegasmust equal the number of atoms in the structure.
-
vec2d externalCharges(const std::vector<std::array<double, 4>> &point_charges, const Structure &structure)
Calculates the one-electron Coulomb integrals with given point charges. Uses OpenMP parallelization.
-
vec2d externalChargesErf(const std::vector<std::array<double, 4>> &point_charges, const std::vector<double> &omegas, const Structure &structure)
Calculates the attenuated one-electron Coulomb integrals with given point charges. Uses OpenMP parallelization.
Important
The numbers of attenuation parameters
omegasand point chargespoint_chargesmust be equal.
-
vec2d externalChargesKernel(size_t ipair, const std::vector<std::array<double, 4>> &charges, const BoysGrid &boys_grid, const ShellPairData &sp_data)
Calculates a batch of one-electron Coulomb integrals for a list of given charges.
for (const lible::ints::ShellPairData &sp_data : sp_data_all) { // The boys grid must be initialized with the angular momentum correct angular momentum: auto [la, lb] = sp_data.getLPair(); int lab = la + lb; lible::ints::BoysGrid boys_grid(lab); for (size_t ipair = 0; ipair < sp_data.n_pairs_; ipair++) { lible::vec2d ints_batch = lible::ints::externalChargesKernel( ipair, charges, boys_grid, sp_data); // Consume the integrals.. } }
-
vec2d externalChargesErfKernel(size_t ipair, const std::vector<std::array<double, 4>> &charges, const std::vector<double> &omegas, const BoysGrid &boys_grid, const ShellPairData &sp_data)
Calculates a batch of attenuated Coulomb integrals.
Important
The numbers of attenuation parameters
omegasand chargeschargesmust be equal.
-
std::array<vec2d, 6> externalChargesD1Kernel(size_t ipair, const std::vector<std::array<double, 4>> &charges, const BoysGrid &boys_grid, const ShellPairData &sp_data)
Calculates a batch of first-derivative one-electron Coulomb integrals.
Important
Due to differentiation, the Boys function grid has to be initialized with total angular momentum incremented by one:
auto [la, lb] = sp_data.getLPair(); int lab1 = la + lb + 1; lible::ints::BoysGrid boys_grid(lab1);
-
std::array<vec2d, 6> externalChargesErfD1Kernel(size_t ipair, const std::vector<std::array<double, 4>> &charges, const std::vector<double> &omegas, const BoysGrid &boys_grid, const ShellPairData &sp_data)
Calculates a batch of first-derivative attenuated one-electron Coulomb integrals.
Important
Due to differentiation, the Boys function grid has to be initialized with total angular momentum incremented by one:
auto [la, lb] = sp_data.getLPair(); int lab1 = la + lb + 1; lible::ints::BoysGrid boys_grid(lab1);
Important
The number of the attenuation parameters
omegasmust equal the number of charges, \((x, y, z, q)\),charges.
-
std::vector<std::array<vec2d, 3>> externalChargesOperatorD1Kernel(size_t ipair, const std::vector<std::array<double, 4>> &charges, const BoysGrid &boys_grid, const ShellPairData &sp_data)
Calculates a batch of one-electron Coulomb operator derivative integrals. The returned list of integrals has the length of the given charges,
charges.
-
std::vector<std::array<vec2d, 3>> externalChargesOperatorErfD1Kernel(size_t ipair, const std::vector<std::array<double, 4>> &charges, const std::vector<double> &omegas, const BoysGrid &boys_grid, const ShellPairData &sp_data)
Calculates a batch of attenuated one-electron Coulomb operator derivative integrals. The returned list of integrals has the length of the given charges,
charges.Important
The Boys function grid has to be initialized with total angular momentum of \(l = l_a + l_b + 1\).
Important
The number of the attenuation parameters
omegasmust equal the number of charges, \((x, y, z, q)\),charges.
-
std::vector<vec2d> potentialAtExternalChargesKernel(size_t ipair, const std::vector<std::array<double, 4>> &charges, const BoysGrid &boys_grid, const ShellPairData &sp_data)
Calculates a batch of one-electron Coulomb integrals at the given charges, \((x, y, z, q)\). Returns a list of integrals with the length of given charges.
-
std::vector<vec2d> potentialAtExternalChargesErfKernel(size_t ipair, const std::vector<std::array<double, 4>> &charges, const std::vector<double> &omegas, const BoysGrid &boys_grid, const ShellPairData &sp_data)
Calculates a batch of attenuated one-electron Coulomb integrals at the given charges, \((x, y, z, q)\). Returns a list of integrals with the length of given charges.
Important
The list of charges must have the same length as the attenuated parameters (
omegas).
-
std::array<vec2d, 3> dipoleMoment(const std::array<double, 3> &origin, const Structure &structure)
Calculates dipole moment integrals. Uses OpenMP parallelization.
-
std::array<vec2d, 3> dipoleMomentKernel(size_t ipair, const std::array<double, 3> &origin, const ShellPairData &sp_data)
Calculates a batch of dipole moment integrals.
-
std::array<vec2d, 3> spinOrbitCoupling1El(const Structure &structure)
Calculates spin-orbit coupling (SOC) one-electron integrals. Uses OpenMP parallelization.
-
std::array<vec2d, 3> spinOrbitCoupling1ElKernel(size_t ipair, const std::vector<std::array<double, 4>> &charges, const BoysGrid &boys_grid, const ShellPairData &sp_data)
Calculates a batch of of spin-orbit coupling (SOC) one-electron integrals.
Important
The boys grid must be initialized with \(l = l_a + l_b + 1\).
-
std::array<vec2d, 3> momentum(const Structure &structure)
Calculates linear momentum integrals. Uses OpenMP parallelization.
-
std::array<vec2d, 3> momentumKernel(size_t ipair, const ShellPairData &sp_data)
Calculates a batch of linear momentum integrals.
-
std::array<vec2d, 3> angularMomentum(const std::array<double, 3> &origin, const Structure &structure)
Calculates angular momentum integrals. Uses OpenMP parallelization.
-
std::array<vec2d, 3> angularMomentumKernel(size_t ipair, const std::array<double, 3> &origin, const ShellPairData &sp_data)
Calculates a batch of angular momentum integrals.
-
arr2d<vec2d, 3, 3> pVpIntegrals(const Structure &structure)
Calculates the \(\hat{p}_i \hat{V}\hat{p}_j\) integrals. Used in X2C: https://doi.org/10.1063/1.4803693. Uses OpenMP parallelization.
-
arr2d<vec2d, 3, 3> pVpKernel(size_t ipair, const std::vector<std::array<double, 4>> &charges, const BoysGrid &boys_grid, const ShellPairData &sp_data)
Calculates a batch of \(\hat{p}_i \hat{V}\hat{p}_j\) integrals.
Important
The boys grid must be initialized with \(l = l_a + l_b + 2\).
-
std::vector<double> eri2Diagonal(const Structure &structure)
Calculates the diagonal part of the two-center Coulomb repulsion integrals in auxiliary basis: \((K|K)\). Uses OpenMP parallelization.
Important
The
structuremust be initialized with an auxiliary basis set beforehand.
-
vec2d eri4Diagonal(const Structure &structure)
Calculates the diagonal part of the four-center Coulomb repulsion integrals, \((\mu\nu|\mu\nu)\). Uses OpenMP parallelization.
-
vec3d eri3(const Structure &structure)
Calculates the three-center Coulomb repulsion integrals, \((\mu\nu|K)\). Uses OpenMP parallelization.
-
vec4d eri4(const Structure &structure)
Calculates the four-center Coulomb repulsion integrals, \((\mu\nu|\kappa\tau)\). Uses OpenMP parallelization
-
BasisAtom basisForAtom(int atomic_nr, const std::string &basis_set)
Returns the main basis set for for an atom.
-
BasisAtom basisForAtomAux(int atomic_nr, const std::string &aux_basis_set)
Returns the auxiliary basis set for an atom.
-
basis_atoms_t basisForAtoms(const std::vector<int> &atomic_nrs, const std::string &basis_set)
Returns the main basis set for the given atoms.
-
basis_atoms_t basisForAtomsAux(const std::vector<int> &atomic_nrs, const std::string &aux_basis_set)
Returns the auxiliary basis set for the given atoms.
-
std::vector<std::tuple<int, int, double>> sphericalTrafo(int l)
Returns the Cartesian to spherical transformation for the given angular momentum.
std::vector<double> atomic_orbitals_cart(lible::ints::numCartesians(l)); // Calculate values of atomic orbitals auto spherical_trafo = lible::ints::sphericalTrafo(l); std::vector<double> atomic_orbitals_sph(lible::ints::numSphericals(l)); // Transform AOs in Cartesian basis to spherical basis. for (const auto& [mu, mu_, val] : spherical_trafo) atomic_orbitals_sph[mu] += val * atomic_orbitals_cart[mu_];
<lible/ints/shell.hpp>
-
struct BasisShell
Structure representing the basis set for an atomic orbital shell.
-
int l_
Angular momentum of the shell.
-
std::vector<double> exps_
Gaussian primitive exponents.
-
std::vector<double> coeffs_
Contraction coefficients of the Gaussian primitives.
-
int l_
-
using basis_shells_t = std::vector<BasisShell>
Type for representing basis sets on a list of shells.
-
struct BasisAtom
Structure representing the basis set on an atom.
-
int atomic_nr_
Atomic number of the atom.
-
basis_shells_t basis_shells_
List of shell basis sets on the atom.
-
int atomic_nr_
-
using basis_atoms_t = std::vector<BasisAtom>
Type representing a list of basis sets on atoms. Object of this type can be used to represent the entire molecular basis set (main or auxiliary).
-
struct Shell
Structure representing an atomic orbital shell. Contains essential data for calculating integrals. rambleramble.
-
int l_
Angular momentum of the shell.
-
int z_
Atomic number of the shell’s atom.
-
size_t dim_cart_
Number of Cartesian atomic orbitals.
-
size_t dim_sph_
Number of spherical atomic orbitals.
-
size_t ofs_cart_
Starting position of the current shell’s Cartesian atomic orbitals in the list of all AOs.
-
size_t ofs_sph_
Starting position of the current shell’s spherical atomic orbitals in the list of all AOs.
-
size_t idx_
Index of the current shell in the list of all shells.
-
size_t idx_atom_
Index of the current shell’s atom in the list of all atoms.
-
std::array<double, 3> xyz_coords_
Cartesian coordinates of the shell’s atom.
-
std::vector<double> exps_
Gaussian primitive exponents.
-
std::vector<double> coeffs_
Contraction coefficients of the Gaussian primitives.
-
std::vector<double> norms_
Normalization coefficients of the spherical atomic orbitals.
-
std::vector<double> norms_prim_
Normalization coefficients of the Gaussian primitives.
-
int l_
<lible/ints/shell_pair_data.hpp>
-
struct ShellData
Structure containing flattened contiguous data of shells with equal angular momentum. Mainly for calculating integrals involving auxiliary basis, e.g. \((K|L)\) and \((\mu\nu|K)\).
An arbitrary vector of shells can be converted into
ShellDataas followsstd::vector<lible::ints::Shell> shells = lible::ints::constructShells( basis_atoms, coords_atoms); std::vector<lible::ints::ShellData> sh_data = lible::ints::shellData(shells);
The shell data is arranged into the vector by different angular momentum. Alternatively, shell data for auxiliary basis can be created directly from the molecular structure:
std::vector<lible::ints::Shell> sh_data = lible::ints::shellDataAux(structure);
-
ShellData(int l, std::vector<Shell> &shells);
Constructs the shell data from the given
shells. The angular momentum of the shells must equall. Cannot be called inside an OMP parallel region.
-
int l_
Angular momentum of the shells.
-
size_t n_shells_
Number of shells.
-
size_t n_primitives_
Total number of primitive Gaussians from all the shells.
-
std::vector<double> coeffs_
Contraction coefficients with Gaussian primitive norms multiplied into.
-
std::vector<double> exps_
Exponents of the primitive Gaussians.
-
std::vector<double> norms_
Normalization constants of the spherical atomic orbitals.
-
std::vector<size> atomic_idxs_
Indices of the atoms involved in the shells.
-
std::vector<size_t> cdepths_
Contraction depth for each shell.
-
std::vector<size_t> coffsets_
Offsets of the contraction coefficients and exponents for each shell.
-
std::vector<size_t> offsets_ecoeffs_
Offsets of the spherical Hermite expansion coefficients for each shell.
-
std::vector<size_t> offsets_norms_
Offsets of the atomic orbital normalization constants for each shell.
-
std::vector<size_t> offsets_sph_
Offsets of the atomic orbital positions in the list of all atomic orbitals.
-
std::vector<size_t> shell_idxs_
Indidices of the involved shells in the list of all shells.
-
ShellData(int l, std::vector<Shell> &shells);
-
struct ShellPairData
Structure containing flattened contiguous data of shell pairs. Main intermediate data structure suitable for calculating most of the one-and two-electron integrals. The shell pair data can be constructed from two lists of arbitrary shells:
// Get the shells: std::vector<lible::ints::Shell> shells_a; std::vector<lible::ints::Shell> shells_b; std::vector<lible::ints::ShellPairData> sp_data = lible::ints:shellPairData( shells_a, shells_b);
The returned shell pair data
sp_datacontains shell pair data for all at the angular momentum pairs. Similar toShellData, the shell pair data can be conveniently obtained from// Get the structure: lible::ints::Structure structure; std::vector<lible::ints::ShellPairData> sp_data = lible::ints::shellPairData( false, structure);
Note that the above given flag implies that symmetry is not used.
-
ShellPairData(bool use_symm, int la, int lb, const std::vector<Shell> &shells_a, const std::vector<Shell> &shells_b, double primitives_thrs = 1e-15);
Using the flag
use_symmtrue forcesishell_a >= ishell_bwhenla == lb. For lists of shells with unequal angular momentum, the symmetry can be made use of by forcingla > lb.The primitives are screened based on the pre-exponential factor for two Gaussian primitives:
\[K_{ab} = \exp(-\mu R^2_{AB})\]where \(R_{AB}\) is the distance between the two Gaussian centers and
\[\mu = \frac{ab}{a + b}\]with \(a\) and \(b\) being the Gaussian primitive exponents. In order to disable screening, set
primitives_thrsto zero.
-
bool uses_symm_
Flag indicating whether symmetry was used or not.
-
double primitive_thrs_
Threshold for screening the primitive Gaussian pairs.
-
int la_
Angular momentum in the first shell.
-
int lb_
Angular momentum in the second shell.
-
size_t n_pairs_
Number of shell pairs (after screening).
-
size_t n_pairs_total_
Total number of shell pairs.
-
size_t n_ppairs_total_
Total number of primitive Gaussian pairs.
-
std::vector<size_t> nrs_ppairs_
Number of Gaussian primitive pairs for each shell pair.
-
std::vector<size_t> offsets_primitives_
Offsets of the Gaussian primitives for each shell pair.
-
std::vector<size_t> offsets_sph_
Offsets of the spherical atomic orbitals in the list of all atomic orbitals.
-
std::vector<size_t> offsets_cart_
Offsets of the Cartesian atomic orbitals in the list of all (Cartesian) atomic orbbitals.
-
std::vector<size_t> offsets_norms_
Offsets of the shell (in a shell pair) atomic orbital norms.
-
std::vector<size_t> offsets_ecoeffs_
Offsets of the spherical Hermite expansion coefficients.
-
std::vector<size_t> offsets_ecoeffs_deriv1_
Offsets of the 1st derivative spherical Hermite expansion coefficients.
-
std::vector<size_t> offsets_ecoeffs_deriv2_
Offsets of the 2nd derivative spherical Hermite expansion coefficients.
-
std::vector<size_t> atomic_idxs_
Indices of atoms involved in the shell pairs.
-
std::vector<size_t> shell_idxs_
Indices of the involved shells in the list of all shells.
-
ShellPairData(bool use_symm, int la, int lb, const std::vector<Shell> &shells_a, const std::vector<Shell> &shells_b, double primitives_thrs = 1e-15);